Data Analysis - Optimization & Performance Improvement
Even though I have a complete analysis pipeline, there are still more things to do to improve the throughput.
This section includes four components: file structure, processing gz in parallel, multithreading, and prefetching data.

File Structure
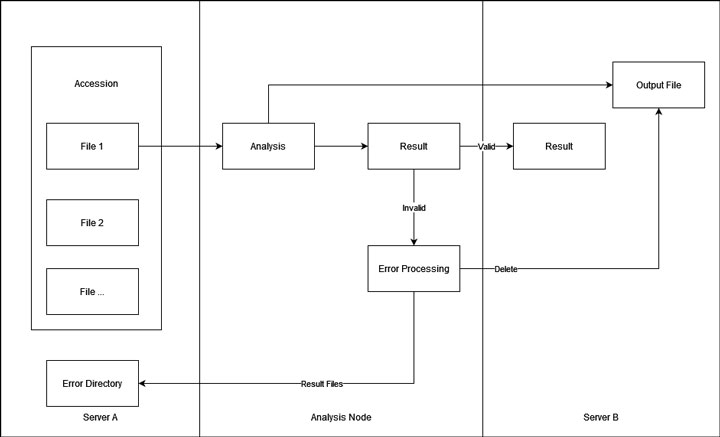
Given that we want to store the data and result with an optimal performance, I choose to put them on a separate server. That is, the file will be first fetched from storage server A, processed, and then the final result to storage server B. This will avoid reading and writing data on the same HDD array as much as possible.
In addition, the size and number of files that each accession contains can vary a lot. Transferring those files could be time-consuming and require a lot of space. Some of the accession is even several TBs large. Therefore, I decided to fetch one file at a time for each job. Also, the output file (except for the results) will be put on server B as well. If the result is valid, the file on server B will be moved to another location on the same server instantly and waiting for the storage pipeline. If the result is invalid, the file on server B will be deleted.
Processing gz in Parallel
Performance Test
The software we used supports both .fastq.gz and .fastq files. I'm interested in finding out whether the performance could be different for different formats of the same file. If using a plain file format is indeed faster, I might be able to do it before the analysis pipeline at a better efficiency. To be short, processing the same file in plain .fastq format does not provide better performance. For some reason, the performance is even worse. The program seems to be able to process .gz file on the fly. Therefore, I choose to stay with the original .fastq.gz format.
Make it Faster
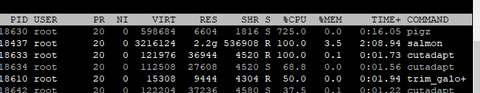
Some software we used (like fastqc) seems to support pigz when possible. pigz is a multithread version of gz, therefore, I have configured pigz for all analysis servers.
Multithreading
Not only pigz, but other components of the pipeline support multithreading as well. Our analysis server has 104 logical cores, and we have to use multithreading when possible.
fastqc
By the time the server was configured, fastqc only supports up to 7 threads per task. We can configure more, but it warns about bad output. The threads for fastqc are then set to 7.
Number of Threads per Job
Components other than fastqc support more than 7 threads, but it is very hard to schedule the cores especially when there are tens of tasks running at the same time. To avoid overloading the server, and for simplicity as well, I choose to use 7 threads for all components of the pipeline.
Server Utilization
We are using 7 threads per job. In order to fully utilize the server, we have to run multiple jobs at the same time. If we run 15 jobs at the same time, we will be using 7 * 15 = 105 threads. This seems to be the right amount. However, after testing, I found that tasks at different stages can not always utilize all 7 threads even if it was configured to do so. Therefore, I slightly over-allocated the resources, and 17 tasks to be run at the same time. Once configured, the server utilization stables at about 95%.
Structure
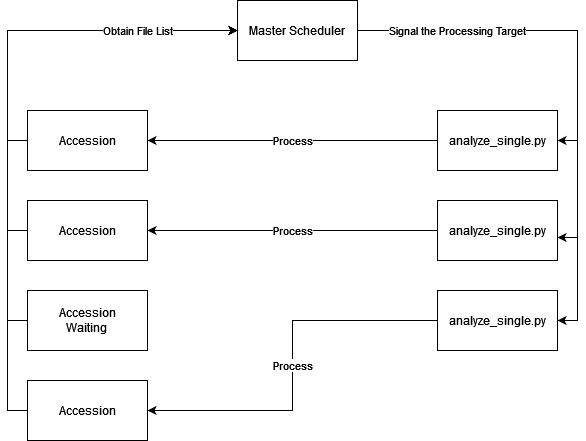
To reduce complexity and also provide better reliability, I developed an analysis script that can handle only one task at a time and a master scheduler that invokes instances of the analysis script targeting different files.
Even though each file within the same accession is processed independently, I decided to run 17 different accessions at the same time, instead of 17 files from a single accession. This is because an accession could contain less than 17 files, which will result in a waste of computing resources. If I run 17 different accessions together with a master scheduler, the CPU is always fully utilized and the overall progress will be optimal, even if it could be slower for a single accession.
The downside of running 17 accessions at the same time is that if the number of tasks in the queue is less than 17, there would be some threads idle. However, given that this is a year-long project, the queue shouldn't be empty at most of the time.
Prefetching Data
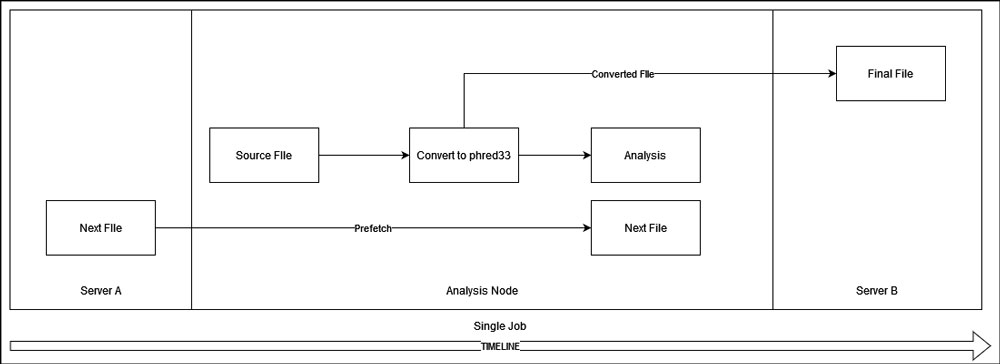
Within the same accession, we analyze each file in a sequence. This will result in resource waste when the server is waiting for the next file to be transferred to localhost. Therefore, I designed a prefetch mechanism. It generally takes a while for each file to be processed, at least longer than the next file to be fetched via a 40G link even if the network is congested. Therefore, the script will try to fetch the next file while the current file is being processed. If the current file is processed and the next file is not here, which is rare, the server will wait for it to be transferred.
In addition, once a file passed its first stage (convert to phred33), the script will start transfering it to server B. The rest steps should take much longer than the transfer task, so the server doesn't have to wait for result transferring and can go directly to the next file after finishing the current one.
Navigate through the Data Analysis Section
- Prev
- Overview
- Software & Structure
- Optimization & Performance Improvement
- Dealing with Errors
- Result Collection
- Automation
- Next